
- 1 Tasks assigned and Questions
- 1.1 Intro To Reference-based Pipeline
- 1.2 Requirements
- 1.3 Explore Experimental Design, Sample Sequence Preview and Reference to Map
- 1.4 Pre-processing (Quality Filtering and Host-Originated Removal)
- 1.5 Taxonomic Profiling with MetaPhlAn2
- 1.6 Functional profiling with HUMAnN and HUMAnN2 Respectively
- 1.7 Code Implementation Log
Original Publish Date: 9th Sep, 2019
Updated on: 09 September, 2019
This Tutorial mainly Originated form Huttenhower Lab Official Reference and Modified by Tank
1 Tasks assigned and Questions
Questions to deal with
Pipeline Numbered Section and Not to Much Explanation 👌
Library Construction Insert Size of Metagenomic Sequencing 👌(300bp/PE150 Novogene and 350/PE100 BIG)
Some Trimmatic Details 👌(reads were removed adaptor sequences, only trimming and qc)
About Orphan Sequence 👌
How Chocophlan Database Generated 👌
(ChocoPhlAn pangenomes A species’ pangenome is a nonredundant representation of the species’ protein-coding potential Using UCLUST, we then cluster all CDSs from high-quality isolate genomes of a given species at 97% nucleotide identity. One representative (centroid) sequence from each cluster is saved. These centroid sequences constitute the species’ pangenome)
- Report Generation with Brief Summary and “what you do with which software and some vital intermediate results” 👌
1.1 Intro To Reference-based Pipeline
This tutorial is set-up to walk you through the process of determining the taxonomic and functional composition of several metagenomic samples. It covers the use of
- Metaphlan2 (for taxonomic classification),
- HUMAnN2 (for functional classification)
1.2 Requirements
- Basic unix skills (there are many tutorials online such as this one)
- Tutorial data (commands to download are below)
- Full Microbiome Helper Virtual Box Image
1.3 Explore Experimental Design, Sample Sequence Preview and Reference to Map
1.3.1 Experimental Design
The first step in any metagenomics pipeline should be to explore your raw files. For this tutorial there should be a “mapping file”, which contains information about each of the samples called map.txt, along with the sequence data for each sample contained within separate FASTQ files within the directory seq
wc -l map.txt
head map.txt| SampleID | groupID | genotype | compartment | timepoint | replicate | batch | techrep | project | indexnum | pheno | library | longtitute |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D6303 | drStrMH | MH | R | NA | 1 | 2 | 2 | E | 41 | stress | 4 | NA |
| D6307 | drStrMH | MH | R | NA | 2 | 2 | 2 | E | 41 | stress | 4 | NA |
| D6314 | drStrMH | MH | R | NA | 3 | 2 | 2 | E | 41 | stress | 4 | NA |
| D6315 | drStrMH | MH | R | NA | 4 | 2 | 2 | E | 41 | stress | 4 | NA |
| DWYJ03 | drStrWY | WYJ | R | NA | 5 | 2 | 2 | E | 41 | stress | 4 | NA |
| DWYJ11 | drStrWY | WYJ | R | NA | 6 | 2 | 2 | E | 41 | stress | 4 | NA |
| DWYJ12 | drStrWY | WYJ | R | NA | 7 | 2 | 2 | E | 41 | stress | 4 | NA |
| DWYJ15 | drStrWY | WYJ | R | NA | 8 | 2 | 2 | E | 41 | stress | 4 | NA |
| W6306 | wetCtrlMH | MH | R | NA | 1 | 2 | 2 | E | 41 | normal | 4 | NA |
| W6308 | wetCtrlMH | MH | R | NA | 2 | 2 | 2 | E | 41 | normal | 4 | NA |
| W6312 | wetCtrlMH | MH | R | NA | 3 | 2 | 2 | E | 41 | normal | 4 | NA |
| WWYJ08 | wetCtrlWY | WYJ | R | NA | 4 | 2 | 2 | E | 41 | normal | 4 | NA |
| WWYJ12 | wetCtrlWY | WYJ | R | NA | 5 | 2 | 2 | E | 41 | normal | 4 | NA |
| WWYJ13 | wetCtrlWY | WYJ | R | NA | 6 | 2 | 2 | E | 41 | normal | 4 | NA |
1.3.2 Sample Sequence Summary Statistics Preview with Fastqc
For your own data you may identify outlier samples with low read depth at this stage. It would also be advisable to explore the quality of your data with a tool like FASTQC.
time fastqc -t 12 seq/D6303_1.fq.gz # about 1m30s
time fastqc -t 12 seq/D6303_2.fq.gz # about 1m30sthen we use the multiqc to aggregate results from bioinformatics analyses across many samples into a single report
time multiqc -d seq/ -o result/see the multiqc aggregated result as belows:multiqc report
for sake of convenience we gunzip the compressed file
gunzip raw_data_example/*gz
wc -l raw_data_example/*.fq ##(stat infor)
1.3.3 Reference Genomes to Map and Indexing-Building
Host Oryza sativa Indica Group and Japonica Group from EnsemblePlants Cultivated rice (O. sativa) and its closest wild relative rice (O. rufipogon)
Note1
Fist to download the corresponding rice genome
wget -c ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/oryza_sativa/dna/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa.gz
wget -c ftp://ftp.ensemblgenomes.org/pub/plants/release-44/fasta/oryza_indica/dna/Oryza_indica.ASM465v1.dna.toplevel.fa.gzfor sake of convenience, here I only concatenate the Jap and Ind v44 into riceJapIndAggreEnselPlts_v44.fa and its aggregated size 779M
cat Oryza_indica.ASM465v1.dna.toplevel.fa Oryza_sativa.IRGSP-1.0.dna.toplevel.fa > riceJapIndAggreEnselPlts_v44.fa
du -sh riceJapIndAggreEnselPlts_v44.fa
for the downstream mapping with bowtie2 so database indexing as follows:
bowtie2-build riceJapIndAggreEnselPlts_v44.fa ensPltV44 ## 1h1.4 Pre-processing (Quality Filtering and Host-Originated Removal)
1.4.1 Quality filtering with Trimmomatic and Host-Originated Removal with Bowtie2
Quality filtering and Screening out of Contaminant Reads were performed by Trimmomatic and Bowtie2 which is aggregated by a helpful wrapper kneadData
time (kneaddata -i raw_data_example/D6303_1.fq -i raw_data_example/D6303_2.fq -o kneaddata_out \
-db /mnt/m1/xiaoning/database/rice/ --trimmomatic /conda/share/trimmomatic-0.39-1/ \
-t 4 --trimmomatic-options "SLIDINGWINDOW:4:20 MINLEN:50" \
--bowtie2-options "--very-sensitive --dovetail" --remove-intermediate-output) >>kneaddata.log 2>&1 ## about 24m you could find the step1 log file
1.4.2 Kneaddata Results Visiualization
You can get a summary of read counts passed at each step broken down by paired and unmatched reads with this command:
kneaddata_read_count_table --input kneaddata_out --output kneaddata_read_counts.txtlibrary(knitr)
library(ggplot2)
library(reshape2)
mapFile <- read.table("./map.txt",row.names= 1, header=T, sep="\t")
kneaddata_read_counts <- read.table("./kneaddata_read_counts.txt",row.names= 1, header=T, sep="\t")
kable(x = kneaddata_read_counts,caption = "kneaddata intermediate result")| raw.pair1 | raw.pair2 | trimmed.pair1 | trimmed.pair2 | trimmed.orphan1 | trimmed.orphan2 | decontaminated.ensPltV44.pair1 | decontaminated.ensPltV44.pair2 | decontaminated.ensPltV44.orphan1 | decontaminated.ensPltV44.orphan2 | final.pair1 | final.pair2 | final.orphan1 | final.orphan2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D6303_1_kneaddata | 3333786 | 3333786 | 2982355 | 2982355 | 176026 | 93315 | 1519310 | 1519310 | 99593 | 65778 | 1519310 | 1519310 | 99593 | 65778 |
index<- melt(kneaddata_read_counts)## No id variables; using all as measure variablesp <- ggplot(index, aes(x=variable, y=value,color=variable)) +
geom_boxplot(alpha=1, outlier.size=0, size=0.7, width=0.4, fill="transparent") +
geom_jitter(aes(shape=variable), position=position_jitter(0.07), size=1, alpha=0.7) +
# scale_colour_manual(values=as.character(colors$color)) +
# scale_shape_manual(values=shapes$shape) +
labs(x="various intermediated read", y="Counts of read by kneaddata") +
theme(axis.text.x = element_text(size=10,angle = 90))
p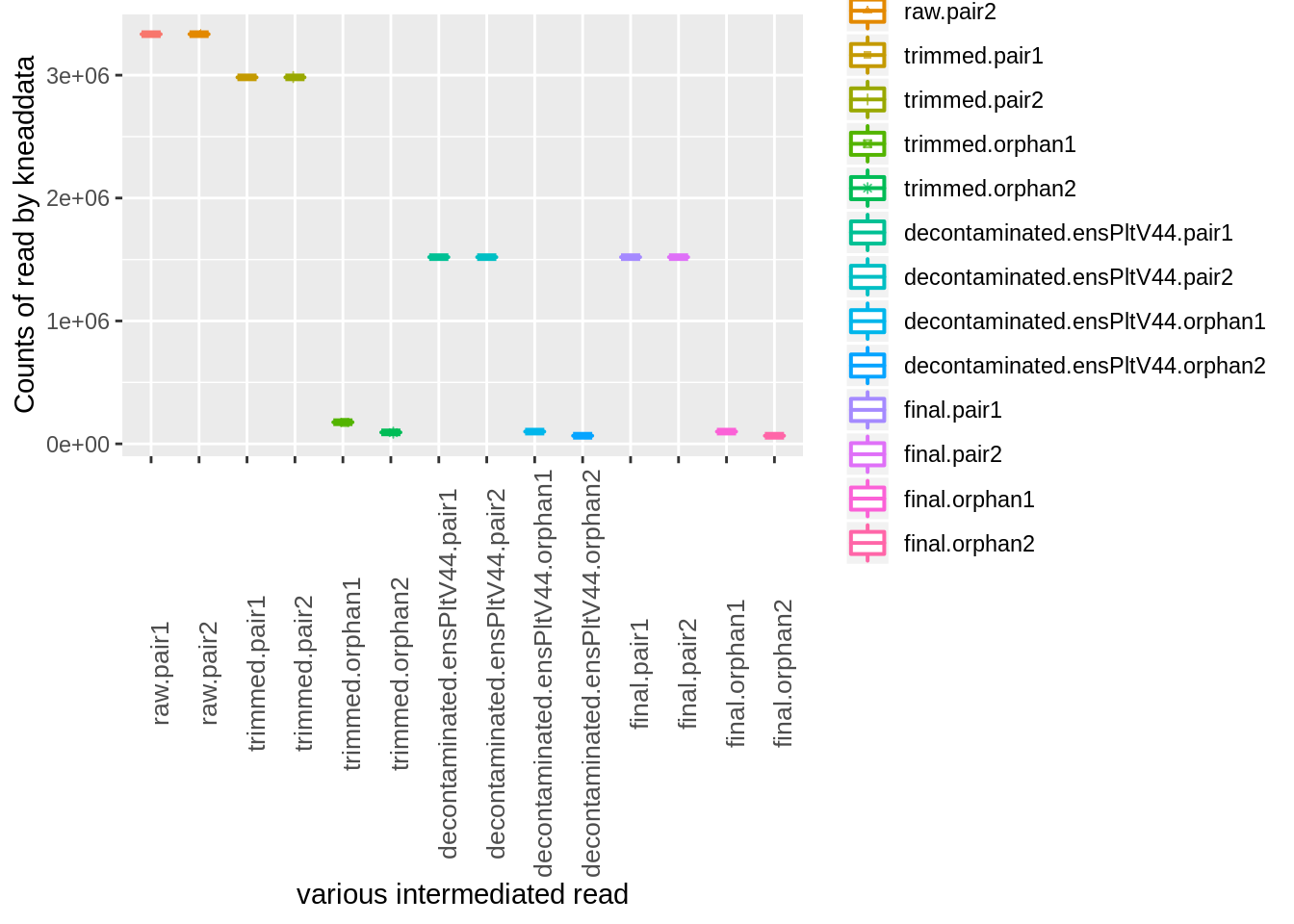
Lastly, We’ll need to move the contaminated sequences and retain read pairs where both forward and reverse reads passed the pre-processing steps
mkdir kneaddata_out/contam_seq
mv kneaddata_out/*_contam_*fastq kneaddata_out/contam_seq
concat_paired_end.pl -p 4 --no_R_match -o cat_reads kneaddata_out/*_paired_*.fastq ## ? parameter question?
1.5 Taxonomic Profiling with MetaPhlAn2
1.5.1 Metaphlan2 Output
The MetaPhlAn2 output file is the “*bugs_list.tsv” in the temp folder within the output folder. You can take a look at it with less.
Your should see something like this:
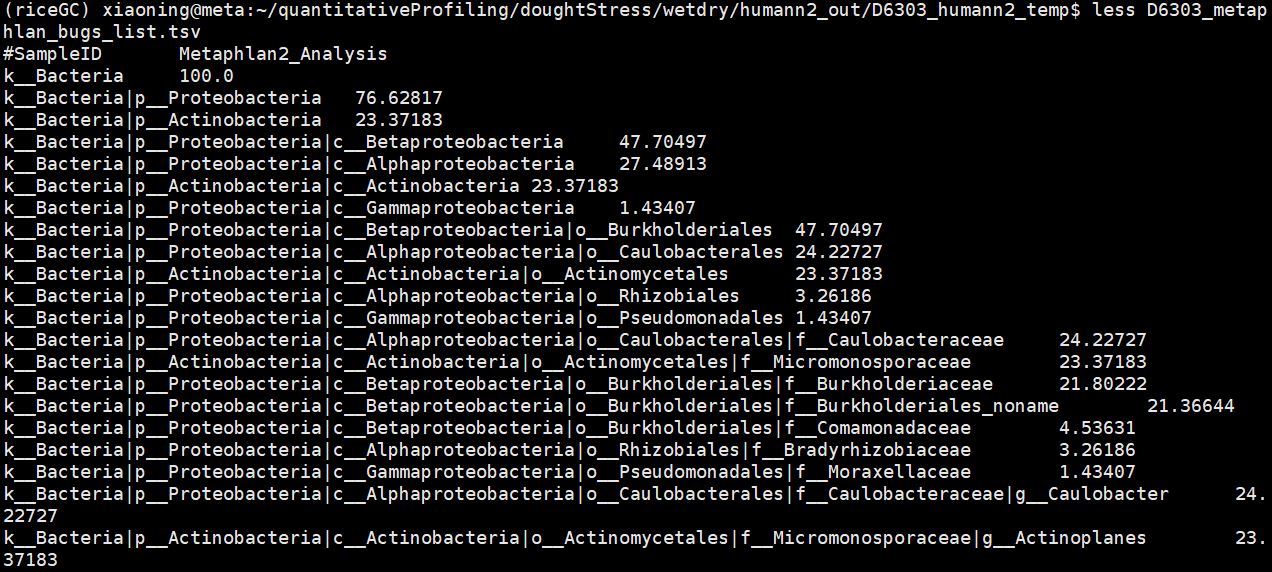
grep 'unclassified' D6303_metaphlan_bugs_list.tsv 1.5.2 Merging Metaphlan2 Results
We’ve placed all of the MetaPhlAn2 output files in precalculated/metaphlan2_out. All of these files can be merged into one table with this command:
merge_metaphlan_tables.py precalculated/metaphlan2_out/*tsv > metaphlan2_merged.tsvTo plot the taxon heatmap:
metaphlan_hclust_heatmap.py --in metaphlan2_merged.tsv \
--out metaphlan2_merged_taxonomy_heatmap_top.pdf \
-c bbcry --top 4 --minv 0.1 -s log
the result with demo as this link pdf
1.6 Functional profiling with HUMAnN and HUMAnN2 Respectively
1.6.1 Running the HUMAnN2 Pipeline (which includes MetaPhlAn2)
1.6.1.1 sigle sample
time (humann2 --threads 8 --input cat_reads/D6303.fastq --output humann2_out/) >>metaPhlAn_HUMAnN.log 2>&1
1.7 Code Implementation Log
1.7.1 Software & Corresponding Dependencies, I/O & Database
- humann2 v0.11.1
- bowtie2 version 2.3
- Output files will be written to: /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out
- Writing temp files to directory: /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp
- Search mode set to uniref90 because a uniref90 translated search database is selected
1.7.2 Run config settings:
1.7.2.1 DATABASE SETTINGS
nucleotide database folder = /home/meta/db/humann2/chocophlan
protein database folder = /db/humann2/uniref
pathways database file 1 = /conda/lib/python2.7/site-packages/humann2/data/pathways/metacyc_reactions_level4ec_only.uniref.bz2
pathways database file 2 = /conda/lib/python2.7/site-packages/humann2/data/pathways/metacyc_pathways_structured_filtered
utility mapping database folder = /db/humann2/utility_mapping
1.7.2.2 RUN MODES
translated search = diamond
threads = 8
1.7.2.3 SEARCH MODE
search mode = uniref90
identity threshold = 90.0
1.7.2.4 ALIGNMENT SETTINGS
evalue threshold = 1.0
prescreen threshold = 0.01
translated subject coverage threshold = 50.0
translated query coverage threshold = 90.0
1.7.2.5 PATHWAYS SETTINGS
minpath = on
xipe = off
gap fill = on
1.7.2.6 INPUT AND OUTPUT FORMATS
input file format = fastq
output file format = tsv
output max decimals = 10
remove stratified output = False
remove column description output = False
log level = DEBUG
- Load pathways database part 1: /conda/lib/python2.7/site-packages/humann2/data/pathways/metacyc_reactions_level4ec_only.uniref.bz2
- Load pathways database part 2: /conda/lib/python2.7/site-packages/humann2/data/pathways/metacyc_pathways_structured_filtered
1.7.3 Lay-1 Taxonomic prescreen with metaphlan2
- Running metaphlan2.py ……..
- Using software: /conda/bin/metaphlan2.py
/conda/bin/metaphlan2.py /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/cat_reads/D6303.fastq \
-t rel_ab \
--mpa_pkl /conda/bin/db_v20/mpa_v20_m200.pkl \
--bowtie2db /conda/bin/db_v20/mpa_v20_m200 \
-o /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_metaphlan_bugs_list.tsv \
--input_type multifastq \
--bowtie2out /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_metaphlan_bowtie2.txt \
--nproc 8- Total species selected from prescreen: 9
1.7.4 Lay-2 Pangenome search
1.7.4.1 Creating custom ChocoPhlAn database
- Adding file to database: g__Rhodopseudomonas.s__Rhodopseudomonas_palustris.centroids.v0.1.1.ffn.gz
- Adding file to database: g__Caulobacter.s__Caulobacter_vibrioides.centroids.v0.1.1.ffn.gz
Creating custom ChocoPhlAn database
Using software: /bin/gunzip
/bin/gunzip \
-c /home/meta/db/humann2/chocophlan/g__Rhodopseudomonas.s__Rhodopseudomonas_palustris.centroids.v0.1.1.ffn.gz \
/home/meta/db/humann2/chocophlan/g__Caulobacter.s__Caulobacter_vibrioides.centroids.v0.1.1.ffn.gz
- TIMESTAMP: Completed custom database creation: 1 seconds
1.7.4.2 Bowtie2 index buiding and mapping
- Execute command:
/conda/bin/bowtie2-build \
-f /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_custom_chocophlan_database.ffn \
/mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_bowtie2_index
- TIMESTAMP: Completed database index : 69 seconds
- Nucleotide input file is of type: fastq
- Using software: /conda/bin/bowtie2
- Remove file: /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_bowtie2_aligned.sam
- Execute command:
/conda/bin/bowtie2 \
-q \
-x /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_bowtie2_index \
-U /m08/21/2019 11:35:09 PM nt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/cat_reads/D6303.fastq \
-S /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_bowtie2_aligned.sam \
-p 8 \
--very-sensitive- 3038620 reads; of these:0.38% overall alignment rate
- TIMESTAMP: Completed nucleotide alignment: 57 seconds
- Total nucleotide alignments not included based on small percent identities: 3882
- TIMESTAMP: Completed nucleotide alignment post-processing: 85seconds
- Total bugs from nucleotide alignment: 2
- g__Caulobacter.s__Caulobacter_vibrioides: 2337 hits
- g__Rhodopseudomonas.s__Rhodopseudomonas_palustris: 5318 hits
- Total gene families from nucleotide alignment: 2018
- Unaligned reads after nucleotide alignment: 99.7480764294 %
1.7.5 Lay-3 Translated search running diamond
1.7.5.1 Genefamily abundance with diamond
- Remove file: /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_diamond_aligned.tsv
- Running diamond ……..
- Aligning to reference database: uniref90_annotated.1.1.dmnd
- Remove file: /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/diamond_m8_9_XDtR
- Using software: /conda/bin/diamond
- Execute command:
/conda/bin/diamond blastx \
--query /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/D6303_bowtie2_unaligned.fa\
--evalue 1.0 \
--threads 8 \
--max-target-seqs 20 \
--outfmt 6 \
--db /db/humann2/uniref/uniref90_annotated.1.1 \
--out /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/diamond_m8_9_XDtR\
--tmpdir /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6- Execute command:
/bin/cat /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/diamond_m8_9_XDtR- TIMESTAMP: Completed translated alignment post-processing : 1876 seconds
- Total bugs after translated alignment: 3
unclassified: 371720 hits g__Caulobacter.s__Caulobacter_vibrioides: 2337 hits g__Rhodopseudomonas.s__Rhodopseudomonas_palustris: 5318 hits - Total gene families after translated alignment: 24363 - Unaligned reads after translated alignment: 95.8994214479 %
- Computing gene families …
- humann2.quantify.families - DEBUG: Compute gene families
- humann2.store - INFO:
Total gene families : 24363 unclassified : 22494 gene families g__Caulobacter.s__Caulobacter_vibrioides : 827 gene families g__Rhodopseudomonas.s__Rhodopseudomonas_palustris : 1192 gene families
- TIMESTAMP: Completed computing gene families : 61 seconds
1.7.5.2 Pathway abundance and coverage
- Computing pathways abundance and coverage …
- Using python module : /conda/lib/python2.7/site-packages/humann2/quantify/MinPath12hmp.py
- Execute command:
/conda/bin/python /conda/lib/python2.7/site-packages/humann2/quantify/MinPath12hmp.py \
-any /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmppf6IYQ \
-map /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpYsRjNU \
-report /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpZGhVL5 \
-details /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmp69OGKR \
-mps /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpyJYmbY
/conda/bin/python /conda/lib/python2.7/site-packages/humann2/quantify/MinPath12hmp.py \
-any /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpoJMAoh \
-map /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpYsRjNU \
-report /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpyFEcZF \
-details /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpw2N6qI \
-mps /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpSkA6Al
/conda/bin/python /conda/lib/python2.7/site-packages/humann2/quantify/MinPath12hmp.py \
-any /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmp9pjqcS \
-map /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpYsRjNU \
-report /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpHrs0Bq \
-details /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmptCq1Vz \
-mps /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpq3ERBn
/conda/bin/python /conda/lib/python2.7/site-packages/humann2/quantify/MinPath12hmp.py \
-any /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpszWTJj \
-map /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpYsRjNU \
-report /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpZ45ncK \
-details /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpE__fq_ \
-mps /mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_humann2_temp/tmpSqogM6/tmpCQzS3a
Compute pathway abundance for bug: unclassified - Compute pathway abundance for bug: all
- TIMESTAMP: Completed computing pathways : 16 seconds
Output files created:
/mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_genefamilies.tsv
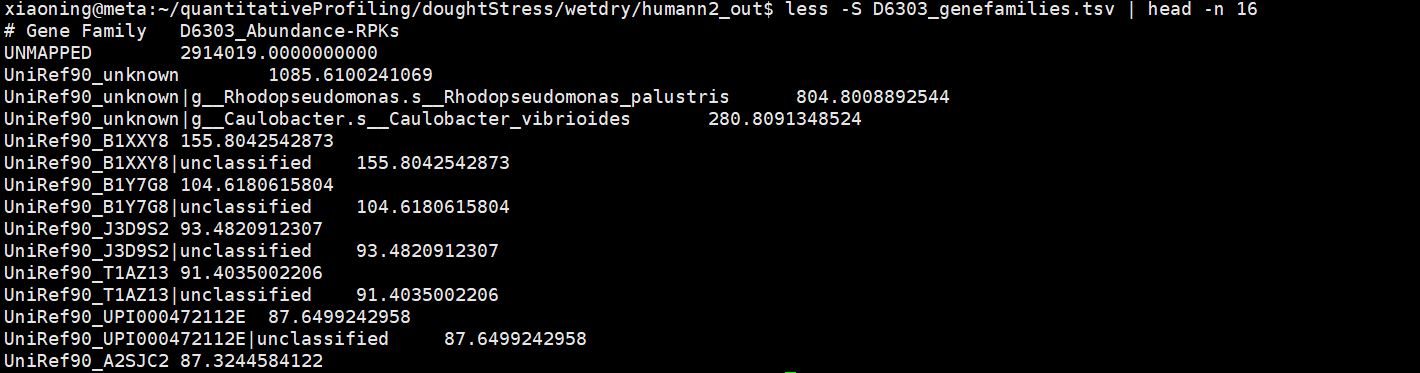
Genefamilies
/mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_pathabundance.tsv
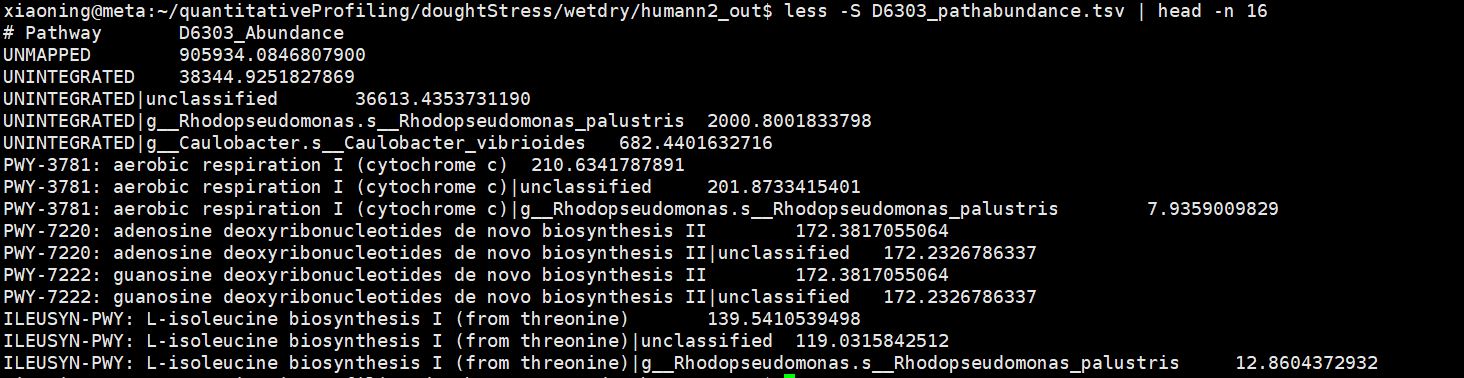
PathwayAbundance
/mnt/m1/xiaoning/quantitativeProfiling/doughtStress/wetdry/humann2_out/D6303_pathcoverage.tsv
PathwayCoverage
previous studies have found that there are functionally important genes that are absent in the reference Nipponbare genome but present in other rice varieties, indicating that one or a few rice genomes cannot include all of the important genomic content. Hence, to comprehensively capture the genomic diversity in rice, it is necessary to de novo construct the complete genomic sequences for dozens of diverse accessions↩
Setting the option –memory-use maximum will speed up the program if you have enough available memory↩